Accelerated Prediction of Phase Behavior for Block Copolymer Libraries Using a Molecularly Informed Field Theory
Charles Li, Elizabeth A. Murphy, Stephen J. Skala, Kris T. Delaney, Craig J. Hawker, M. Scott Shell, Glenn H. Fredrickson
J. Am. Chem. Soc. Oct. 2024
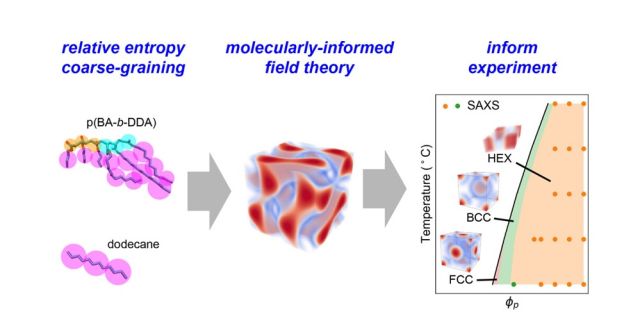
Abstract: Solution formulations involving polymers are the basis for a wide range of products spanning consumer care, therapeutics, lubricants, adhesives, and coatings. These multicomponent systems typically show rich self-assembly and phase behavior that are sensitive to even small changes in chemistry and composition. Longstanding computational efforts have sought techniques for predictive modeling of formulation structure and thermodynamics without experimental guidance, but the challenges of addressing the long time scales and large length scales of self-assembly while maintaining chemical specificity have thwarted the emergence of general approaches. As a consequence, current formulation design remains largely Edisonian. Here, we present a multiscale modeling approach that accurately predicts, without any experimental input, the complete temperature–concentration phase diagram of model diblock polymers in solution, as established postprediction through small-angle X-ray scattering. The methodology employs a strategy whereby atomistic molecular dynamics simulations is used to parametrize coarse-grained field-theoretic models; simulations of the latter then easily surmount long equilibration time scales and enable rigorous determination of solution structures and phase behavior. This systematic and predictive approach, accelerated by access to well-defined block copolymers, has the potential to expedite in silico screening of novel formulations to significantly reduce trial-and-error experimental design and to guide selection of components and compositions across a vast range of applications.